



RESEARCH

Our research focuses on advanced computational modeling to better understand and enhance pulmonary surfactant (PS) functions, particularly regarding their role in human health and drug delivery mechanisms.
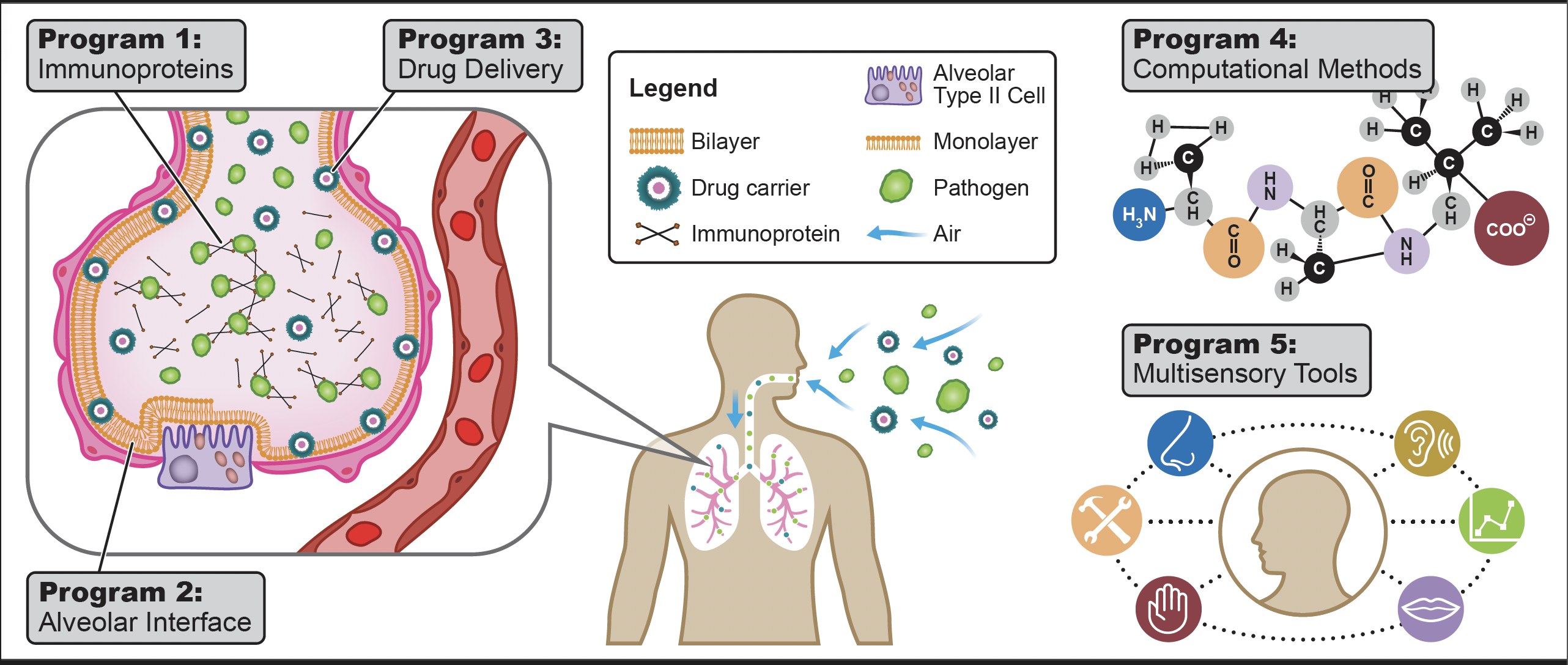
This program explores the structure and function of surfactant proteins such as SP-A, SP-D, and MBL, vital components of the body’s innate immune system. These proteins interact with pathogens by binding to surface glycans, inhibiting their activities. More information about my current research can be found in this poster or on my research group website here: minkaracombinelab.com.
Research in this area aims to understand the monolayer to bilayer transitions of pulmonary surfactant during the breathing process. This involves studying the effects of surfactant proteins (SP-B and SP-C) and various lipids on this transition and how they regulate surface tension, which is crucial for effective lung function.

This program focuses on the micellar and bilayer structures of PS and their potential as drug carriers. Leveraging the lung's characteristics such as high permeability and large surface area, the research aims to develop innovative drug delivery methods that enhance the efficacy of inhalation therapies.
The development and refinement of computational techniques, such as hybrid force fields, are central to this program. These methods allow for efficient modeling of biological systems, integrating both coarse-grained and atomistic approaches to enhance simulation accuracy and effectiveness.
This initiative redefines scientific accessibility by integrating multisensory tools into research, making science comprehensible and inclusive for individuals with sensory impairments. This includes developing tactile graphics and other innovative techniques that provide non-visual ways to interact with scientific data.
In my postdoctoral research, I utilized Monte Carlo algorithms to gain a better understanding of complex chemical systems. More specifically, I explored the interfaces of water/oil mixtures, surfactant mesophases, a new pressure equation of state for chain molecules, methods to determine partial molar properties of mixtures from simulations, and hydrogen/water mixtures at extreme temperatures and pressures. Our research group’s highly specialized Monte Carlo algorithms and simulation protocols allowed for the precise study and prediction of the physical properties of these systems.

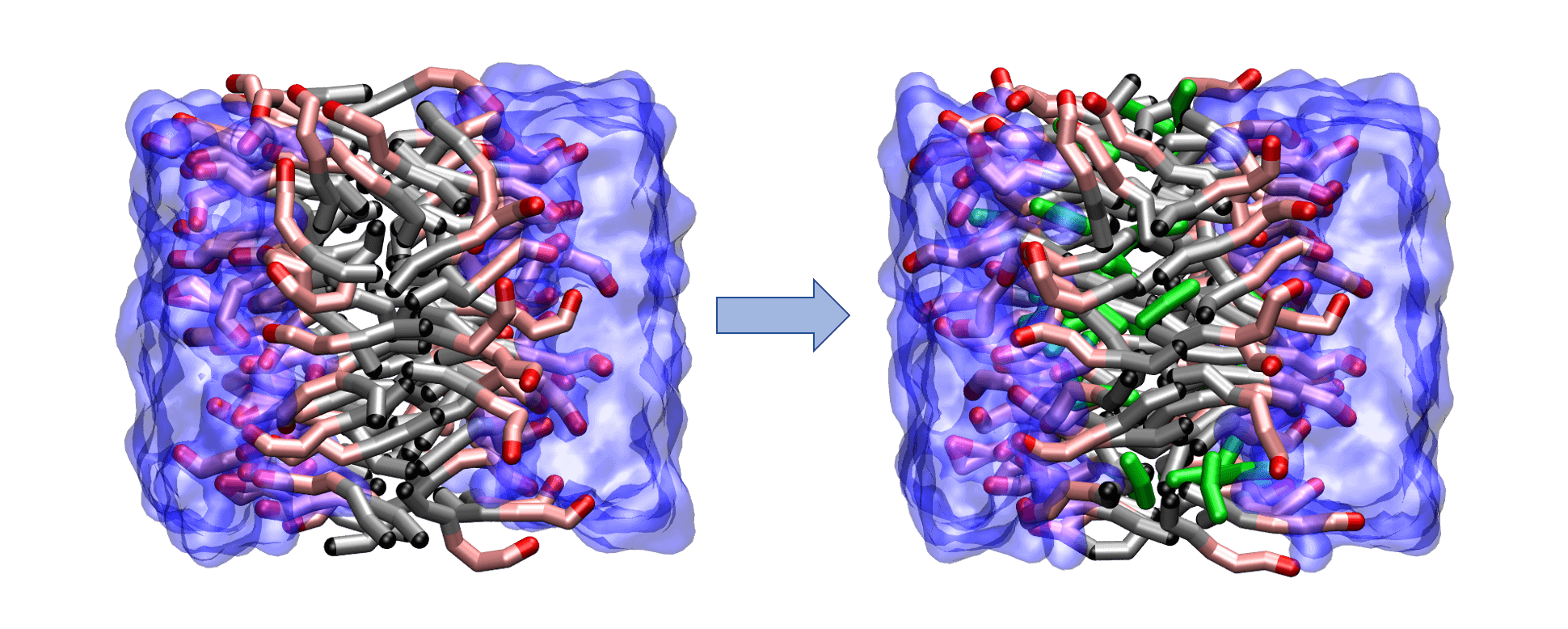
Understanding solute uptake into soft microstructured materials, such as bilayers and worm-like and spherical micelles, is of interest in the pharmaceutical, agricultural, and personal care industries. To obtain molecular-level insight on the effects of solutes loading into a lamellar phase, we utilized the Shinoda-Devan-Klein (SDK) coarse-grained force field in conjunction with configurational-bias Monte Carlo simulations in the osmotic Gibbs ensemble. In these studies, the lamellar phase was comprised of a bilayer formed by triethylene glycol mono-n-decyl ether (C10E3) surfactants surrounded by water with a 50:50 surfactant/water weight ratio. We studied the unary adsorption isotherm and the effects on bilayer structure and stability caused by n-nonane, 1-hexanol, and ethyl butyrate at several different reduced reservoir pressures, as described here. Additionally, we studied the binary adsorption isotherm of n-nonane and 1-hexanol mixtures.

A Langmuir monolayer consists of amphiphilic surfactant molecules that adsorb at a water/air interface to form a unimolecular layer. As well as having multiple applications in templating, chemical sensing, and coatings, these monolayers can serve as a model for lipid bilayers. The precise fluid phase behavior of Langmuir monolayers and other two-dimensional systems is difficult to accurately obtain experimentally. Molecular simulations are needed to probe the vapor-liquid equilibria of these complex systems. In 1994, Siepmann et al. used configurational-bias Monte Carlo simulations in the Gibbs ensemble (CBMC-GE) to study the vapor-liquid coexistence curve (VLCC) of a pentadecanoic acid monolayer, but these simulations were limited to a small system of only 60 surfactant chains and relatively short trajectories. We carried out CBMC-GE simulations for much larger systems and longer trajectories using the TraPPE united-atom force field for the alkyl tails to probe the VLCC of a pentadecanoic monolayer. Furthermore, we investigated modifications to the headgroup-headgroup potential to improve prediction accuracy and to allow extension of the simulations to coexistence of liquid-expanded and liquid-condensed phases. More information is available here.
Knowledge of the interfacial properties of water/oil mixtures is extremely important for the petrochemical industry and for understanding detergency and hydrophobic effects. To probe the liquid/vapor interface of water/n-hexane and water/H2S mixtures, we used configurational-bias Monte Carlo simulations in the osmotic Gibbs ensemble, which allowed us to determine the effects of n-hexane and hydrogen sulfide at several different partial pressures on the liquid/vapor surface tension. By analyzing the simulation trajectories, we were also able to determine the n-hexane and hydrogen sulfide loading, providing key molecular-level insights on the structure of the interfaces. You can watch a presentation I gave at the 2018 Industrial Partnership for Research in Interfacial & Materials Engineering (iPRIME) conference in the video (left column) or read the full article here.
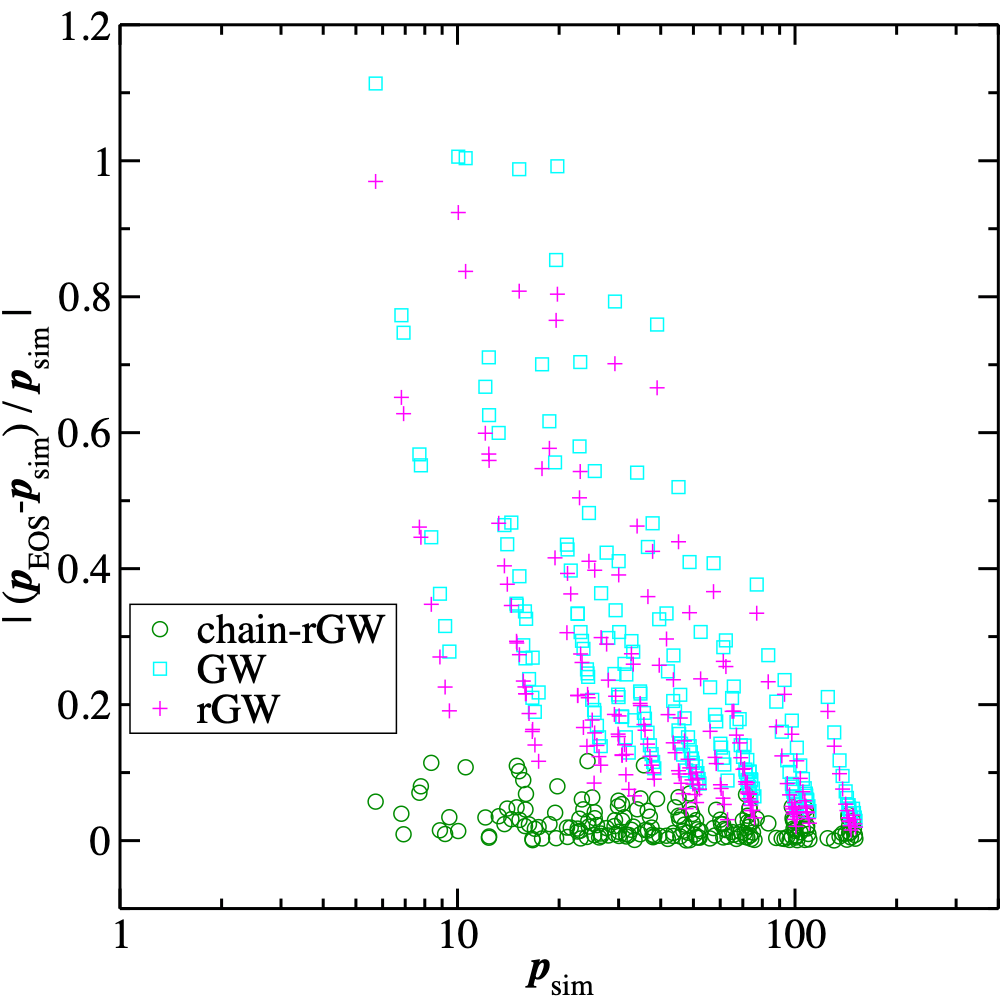
We have developed and tested a chain-revised Groot-Warren equation of state (crGW-EOS) to describe systems of homo-oligomeric chains in the framework of dissipative particle dynamics (DPD). Thermodynamic perturbation theory (TPT) was applied to introduce correction terms that account for the reduction in pressure with an increasing number of bonds at constant bead number density. We modified this equation of state by introducing a set of switching functions that yield an accurate second virial coefficient in the low-density limit. For chain molecules, the crGW-EOS offers several improvements over the revised Groot-Warren equation of state (rGW-EOS) and the original GW-EOS. We tested the crGW-EOS by predicting the pressure of oligomeric systems and the B2 virial coefficient of chain DPD particles for a range of bond lengths. Additionally, we developed a method for determining cross-interaction parameters between chains of different compositions and sizes, for both thermal and athermal mixtures. We also explored how system size affects partitioning near the upper-critical solution temperature. The full article is available here.
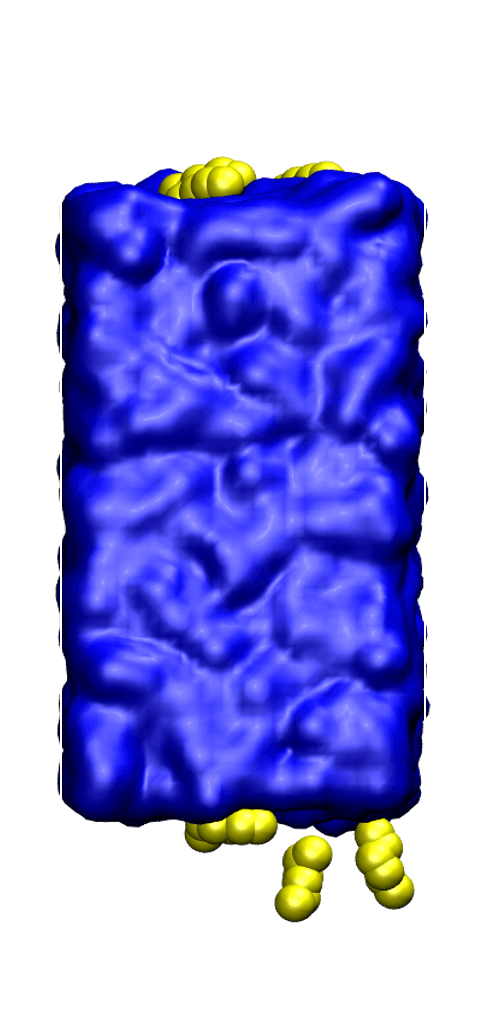
Understanding the effects of surfactants on the structural, transport, and thermodynamic properties of vapor bubbles in aqueous solutions is important for naval research. We used Monte Carlo simulations in the NpT osmotic Gibbs ensemble to study surfactants adsorbing onto liquid/vapor interfaces with different curvature: (i) a vapor bubble in water (with bubble diameters of about 1.7, 2.1, and 2.4 nm), (ii) a free-standing film of water, and (iii) a bulk water phase. The surfactants considered were ethanol and 1-butanol, with water and surfactant molecules modeled using the TIP4P/2005 and TraPPE-UA force fields, respectively. More information on these studies is available in this poster.
Supercritical fluid-fluid phase separations are important for modeling Jovian planets, explaining gas laser phase behavior, and understanding reduction-oxidation chemistry in Earth’s crust and mantle. To explore supercritical fluid-fluid immiscibility of the binary H2O/H2 mixture, we used Monte Carlo simulations in the NpT Gibbs ensemble. We analyzed the atom-atom radial distribution functions for H2O in the H2-rich phase, examined the cluster size distribution of these H2O aggregates, and determined the distribution of H-bond energies and geometries.
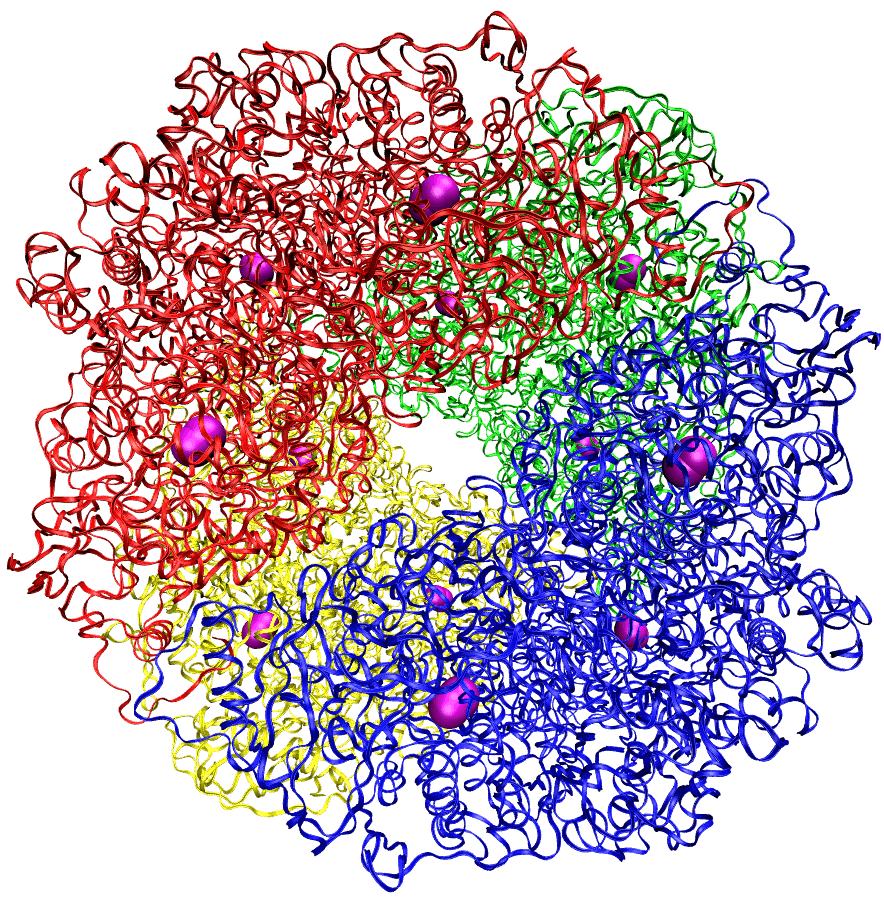
Helicobacter pylori (HP) are gastrointestinal bacteria that infect the majority of the world’s population with no effective cure. These bacteria survive stomach acidity by producing an enzyme, HP urease, that hydrolyzes urea. My Ph.D. projects mostly focused on using molecular dynamics (MD) and molecular docking studies to gain an atomic-level understanding of the structure and function of the enzyme to help develop inhibitors for medical treatment. My thesis is available here. While at the University of Florida, I also conducted quantum calculations to determine the heats of formation for transition metal complexes and the reaction mechanism of amide hydrolysis.
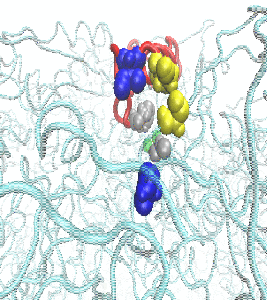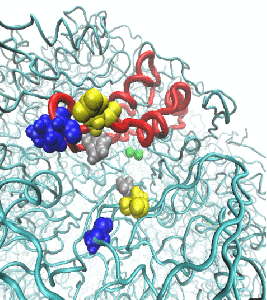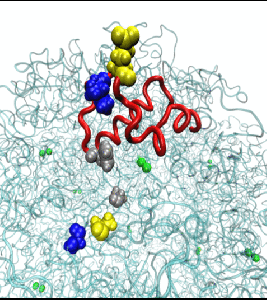
In an effort to determine the mechanism of hydrolysis, we conducted molecular dynamics simulations to study the structural features of the H. pylori urease enzyme in aqueous solution. During these investigations, the flaps that cover the active site cavity of the protein were found to open wider than previously observed, which has significant implications for the design of H. pylori urease inhibitors. More information is available in the publication here.
To investigate the interactions between aqueous ammonium ions (an intermediate byproduct of urea hydrolysis) and H. pylori urease, molecular dynamics simulations were performed on the enzyme in 150 mM aqueous NH4Cl. During these studies, a possible path in which ammonium ions are expelled from the active sites during hydrolysis was observed. The blockage of this pathway was suspected to be a target for enzyme inhibition. A poster of this research is available here.
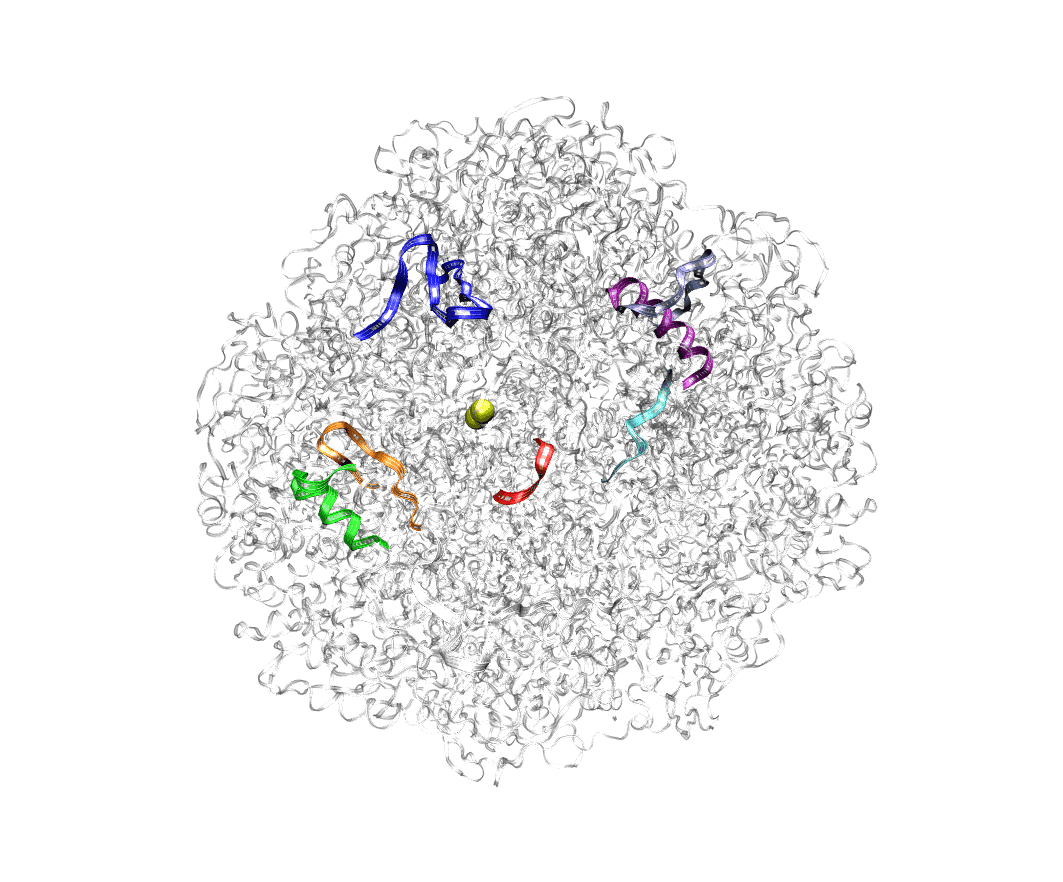
To observe the mechanism of interaction between H. pylori urease and its substrate, urea, molecular dynamics simulations were performed on the urease enzyme in 10.5 M aqueous urea solution. During these simulations, a possible shuttling process of urea into the active site of the protein was observed. These findings allow for the targeting of other regions of the protein that may negatively affect the enzymatic hydrolysis of urea, leading to a greater number of potential inhibitor binding sites. The full text is available here.
While ab initio calculations and density functional theory have been well validated for a wide variety of organic compounds, there is comparably little supporting work that has been done in the area of transition metal-containing complexes. We have used fifteen density functionals and the MP2, CCSD, and CCSD(T) methods with Dunning double and triple-z basis sets, both with and without diffuse functions, in order to determine their utility in predicting the heats of formation and bond lengths of third row transition metal complexes. Preliminary results revealed that the TPSSTPSS and TPSSKCIS density functionals are quite effective in accurately predicting the heat of formation and equilibrium bond lengths for these metal complexes, and that the B3LYP, BLYP, and correlated methods perform much more poorly for the same compounds. These results suggest levels of computational theory that are suitable for applications such as geometry optimizations of metal-containing enzyme active sites, and for serving as the quantum mechanical segment of QM/MM calculations on larger protein structures.
The exact pathway of amide hydrolysis is currently unknown, and experimental and computational data are in some instances inconsistent with one another. In an attempt to elucidate the various mechanistic pathways of urea hydrolysis, we performed electronic structure calculations of the ground states, intermediates, and transition states associated with a variety of possible mechanisms. An array of density functionals were used to characterize these reaction pathways, with the aim of identifying a suitable QM level of theory for studying urease.
We developed a novel computational approach to identify molecules that could reduce urease enzyme activity in H. pylori, K. aerogenes, and many other microorganisms. Using Glide, we docked (a) urea, (b) a 36,000 ligand library, (c) several libraries from the ZINC database including biologically relevant compounds, lead-like structures and drug molecules and finally, (d) selected individual compounds from the literature that had been previously identified as promising inhibitors of urease into the K. aerogenes urease active site. During our investigation, the natural products epigallocatechin and quercetin were shown to inactivate urease by locking the enzyme into the wide-open conformation that we had discovered in our previous investigations. This approach, which focuses on finding compounds that lock an enzyme into a single conformation, can be applied to identify new potential inhibitors for a variety of enzymes. The full publication is available here.

In my undergraduate research at Wellesley College, I used computational component analysis to investigate the electrostatic contributions of chemical moieties in two enzymes with inhibitors: trypsin with bovine pancreatic trypsin inhibitor and HIV-1 reverse transcriptase with several non-nucleoside reverse transcriptase inhibitors. In a separate project, I also investigated the electronic structure of single-walled carbon nanotubes.
I worked to investigate the promiscuity and specificity of the trypsin/bovine pancreatic trypsin inhibitor (BPTI) system through the analysis of the electrostatic component of the free energy, ΔG. By eliminating the charge contribution of each residue on the ligand, we were able to computationally determine the how that residue influenced ΔG, observing that a very positive change in ΔG indicates that that particular residue is important to binding.
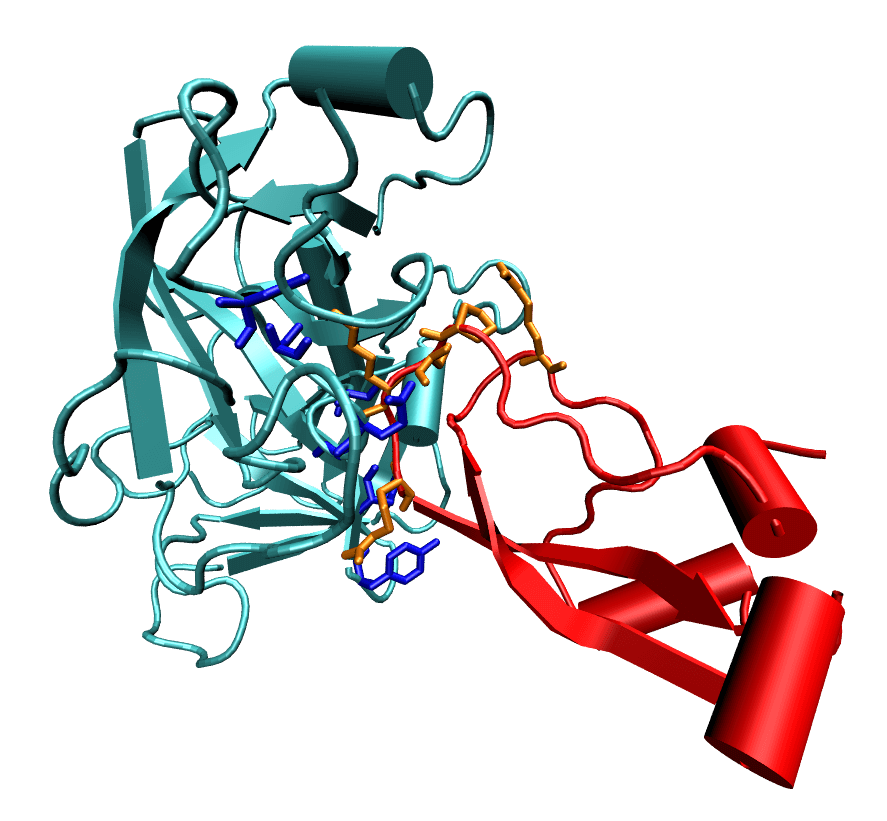
A parallel study was conducted to evaluate the efficiency and accuracy of various numerical methods to determine ΔG electrostatics. The most accurate method, the finite difference method, was compared with the boundary element method, generalized Born, boundary-integral-based electrostatic estimation-Coulomb field approximation, and boundary-integral-based electrostatic estimation-preconditioned methods. The publication is available here.
We conducted a systematic analysis of the electrostatic component of binding of three HIV-1 reverse transcriptase (RT) inhibitors—nevirapine (NVP), efavirenz (EFZ), and rilpivirine (RPV)—to both wild-type (WT) and mutant variants of RT.
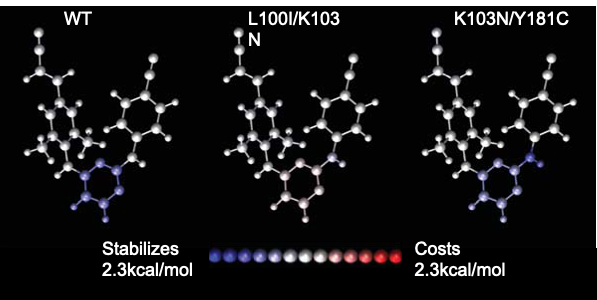
Electrostatic charge optimization was applied to determine how suited each molecule’s charge distribution is for binding WT and individual mutants of HIV-1 RT. We also determined the contributions of chemical moieties on each molecule toward the electrostatic component of binding, and show that different regions of a drug molecule may be used for recognition by different RT variants. The electrostatic contribution of certain RT residues toward drug binding was also computed to highlight critical residues for each interaction. Finally, the charge distribution of RPV is optimized to promiscuously bind to three RT variants rather than to each one in turn, with the resulting charge distribution being a compromise between the optimal charge distributions to each individual variant. This work demonstrates that even in a binding site considered quite hydrophobic, electrostatics play a subtle yet varying role that must be considered in designing next-generation molecules that recognize rapidly-mutating targets. The full publication is available here.
My first research project utilized both semi-empirical PM3 and density functional theory (DFT) B3LYP computations in order to determine the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), and band gap energy patterns of increasing length single-walled carbon nanotubes (SWCNTs).
 Page 4
Page 4